본격적인 공모전 준비에 앞서 홈페이지에 공유되어 있는 코드와 EDA 과정을 분석해보며 아이디어를 구체화시키려 한다.
학습 모델에 대한 분석은 다음 글에서 할 예정이며 이번에는 data EDA를 주로 살펴본다.
1. AMP - EDA + Models
https://www.kaggle.com/code/craigmthomas/amp-eda-models 에 공유되어있는 코드를 분석하려 한다.

1.1 Initial Impressions
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import gc
import warnings
warnings.filterwarnings('ignore')
train_clinical_data = pd.read_csv("../input/amp-parkinsons-disease-progression-prediction/train_clinical_data.csv")
train_peptides = pd.read_csv("../input/amp-parkinsons-disease-progression-prediction/train_peptides.csv")
train_protiens = pd.read_csv("../input/amp-parkinsons-disease-progression-prediction/train_proteins.csv")
supplemental_clinical_data = pd.read_csv("../input/amp-parkinsons-disease-progression-prediction/supplemental_clinical_data.csv")우선 data load와 data EDA에 필요한 라이브러리를 불러온다.
pandas : 데이터 조작 및 분석을 위한 소프트웨어 라이브러리이다. 숫자 테이블과 시계열을 조작하기 위한 데이터 구조와 연산을 제공한다.
numpy : 행렬이나 대규모 다차원 배열을 쉽게 처리할 수 있도록 지원하는 라이브러리이다.
matplotlib : 범용 GUI 툴킷을 사용하여 애플리케이션에 플롯을 포함 하기 위한 객체 지향 API를 제공하는 시각화 라이브러리이다.
seaborn : matplotlib과 같이 시각화 라이브러리로 matplotlib보다 간편하고 디자인이 더 깔끔한 편이다.
gc : garbage collection를 조절하는데 사용하는 라이브러리로 메모리를 사용하지 않을 때, 이를 비우는 기능을 수행한다. Python의 경우 reference cycles를 detect 및 break 할 수 있는 reference counting 과 cyclic garbage collector를 통해 garbage collection을 수행한다.
warnings : 경고 메세지의 생산, 숨김 및 수정을 가능하게 하는 라이브러리이다. 위 코드에서는 경고메세지를 숨김 처리하였다.
이후 저번 블로그에서 기술한 4가지의 train data를 불러와 각각 변수로 지정하였다.
train_clinical_dataprint(": Found {:,d} unique patient_id values".format(train_clinical_data["patient_id"].nunique()))
print(": Found {:,d} unique visit_month values".format(train_clinical_data["visit_month"].nunique()))train_clinical_data에 대해 확인하는 과정이다. 이 코드의 실행 결과로 data의 head와 tail을 확인할 수 있었고, 중복을 제외하여 환자의 id는 248개(이는 환자 248명에 대한 data임을 말한다), 방문 기간의 종류가 17가지 임을 확인할 수 있었다.
train_peptidesprint(": Found {:,d} unique patient_id values".format(train_peptides["patient_id"].nunique()))
print(": Found {:,d} unique UniProt values".format(train_peptides["UniProt"].nunique()))
print(": Found {:,d} unique Peptide values".format(train_peptides["Peptide"].nunique()))위와 마찬가지로 train_peptides에 대한 data를 확인할 수 있었고, 248가지의 환자 id와 227종의 UniProt, 968 가지의 펩타이드 서열을 확인할 수 있었다.
train_protiensprint(": Found {:,d} unique patient_id values".format(train_protiens["patient_id"].nunique()))
print(": Found {:,d} unique visit_month values".format(train_protiens["visit_month"].nunique()))
print(": Found {:,d} unique UniProt values".format(train_protiens["UniProt"].nunique()))train_protiens에 대한 data 확인이며, 248가지의 환자 id와 15가지의 방문 기간, 227종의 UniProt를 확인할 수 있었다.
supplemental_clinical_dataprint(": Found {:,d} unique patient_id values".format(supplemental_clinical_data["patient_id"].nunique()))
print(": Found {:,d} unique visit_month values".format(supplemental_clinical_data["visit_month"].nunique()))supplemental_clinical_data로, 771가지의 환자 id와 8가지의 방문 기간을 확인할 수 있었다.
combined = pd.concat([train_clinical_data, supplemental_clinical_data]).reset_index(drop=True)
combinedtrain_clinical_data와 supplement_clinical_data 두 dataset을 concat 함수로 결합하고, reset_index 함수를 통해 row index를 기본값 0부터 시작하는 순서로 초기화시킨다. 이때, (drop=True)를 통해 기존의 index를 출력되지 않게 하였다.
print(": Found {:,d} unique patient_id values".format(combined["patient_id"].nunique()))
print(": Found {:,d} unique visit_month values".format(combined["visit_month"].nunique()))앞서 4가지 data에 대한 정보를 확인한 것과 같은 방법으로 병합된 data를 확인한 결과 1,019개의 환자 id와 18가지의 방문기간을 확인할 수 있었다.
해당 페이지에서 언급하는 주요 관측을 통해 해당 데이터의 용량이 크지 않으므로 메모리 부담이 적다는 것을 알 수 있었다.
1.2 Null Values

train_clinical_data["null_count"] = train_clinical_data.isnull().sum(axis=1)
counts_train_clinical_data = train_clinical_data.groupby("null_count")["visit_id"].count().to_dict()
null_train_clinical_data = {"{} Null Value(s)".format(k) : v for k, v in counts_train_clinical_data.items()}
train_peptides["null_count"] = train_peptides.isnull().sum(axis=1)
counts_train_peptides = train_peptides.groupby("null_count")["visit_id"].count().to_dict()
null_train_peptides = {"{} Null Value(s)".format(k) : v for k, v in counts_train_peptides.items()}
train_protiens["null_count"] = train_protiens.isnull().sum(axis=1)
counts_train_protiens = train_protiens.groupby("null_count")["visit_id"].count().to_dict()
null_train_protiens = {"{} Null Value(s)".format(k) : v for k, v in counts_train_protiens.items()}
supplemental_clinical_data["null_count"] = supplemental_clinical_data.isnull().sum(axis=1)
counts_supplemental_clinical_data = supplemental_clinical_data.groupby("null_count")["visit_id"].count().to_dict()
null_supplemental_clinical_data = {"{} Null Value(s)".format(k) : v for k, v in counts_supplemental_clinical_data.items()}
fig, axs = plt.subplots(nrows=2, ncols=2, figsize=(15, 15))
axs = axs.flatten()
_ = axs[0].pie(
x=list(null_train_clinical_data.values()),
autopct=lambda x: "{:,.0f} = {:.2f}%".format(x * sum(null_train_clinical_data.values())/100, x),
explode=[0.05] * len(null_train_clinical_data.keys()),
labels=null_train_clinical_data.keys(),
colors=sns.color_palette("Set2")[0:len(null_train_clinical_data.keys())],
)
_ = axs[0].set_title("Null Values Per Row in Clinical Data", fontsize=15)
_ = axs[3].pie(
x=list(null_train_peptides.values()),
autopct=lambda x: "{:,.0f} = {:.2f}%".format(x * sum(null_train_peptides.values())/100, x),
explode=[0.05] * len(null_train_peptides.keys()),
labels=null_train_peptides.keys(),
colors=sns.color_palette("Set2")[0:len(null_train_peptides.keys())],
)
_ = axs[3].set_title("Null Values Per Row in Peptide Data", fontsize=15)
_ = axs[2].pie(
x=list(null_train_protiens.values()),
autopct=lambda x: "{:,.0f} = {:.2f}%".format(x * sum(null_train_protiens.values())/100, x),
explode=[0.05] * len(null_train_protiens.keys()),
labels=null_train_protiens.keys(),
colors=sns.color_palette("Set2")[0:len(null_train_protiens.keys())],
)
_ = axs[2].set_title("Null Values Per Row in Protein Data", fontsize=15)
_ = axs[1].pie(
x=list(null_supplemental_clinical_data.values()),
autopct=lambda x: "{:,.0f} = {:.2f}%".format(x * sum(null_supplemental_clinical_data.values())/100, x),
explode=[0.05] * len(null_supplemental_clinical_data.keys()),
labels=null_supplemental_clinical_data.keys(),
colors=sns.color_palette("Set2")[0:len(null_supplemental_clinical_data.keys())],
)
_ = axs[1].set_title("Null Values Per Row in Supplemental Clinical Data", fontsize=15)해당 시각화 테이블 형성 코드
해당 테이블을 통해 펩타이드와 단백질 데이터로 CSF를 관측할 때 null values가 없다는 것을 알 수 있으며, 임상 데이터에는 null values가 존재함을 확인할 수 있었다.
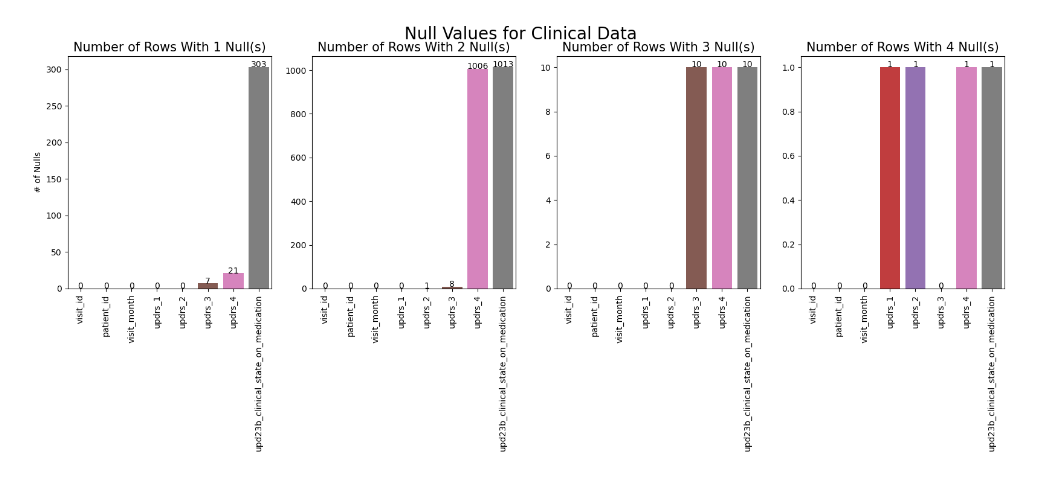
null_count_labels = [train_clinical_data[(train_clinical_data["null_count"] == x)].isnull().sum().index[:-1] for x in range(1, 6)]
null_count_values = [train_clinical_data[(train_clinical_data["null_count"] == x)].isnull().sum().values[:-1] for x in range(1, 6)]
fig, axs = plt.subplots(nrows=1, ncols=4, figsize=(20, 5))
fig.suptitle("Null Values for Clinical Data", fontsize=20)
axs = axs.flatten()
for x in range(0, 4):
ax = axs[x]
labels = null_count_labels[x]
_ = sns.barplot(x=labels, y=null_count_values[x], ax=ax)
_ = ax.set_title("Number of Rows With {} Null(s)".format(x + 1), fontsize=15)
_ = ax.set_ylabel("# of Nulls" if x == 0 else "")
_ = ax.set_xlabel("")
_ = ax.set_xticks([z for z in range(len(labels))], labels, rotation=90)
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:d}".format(int(height)), ha="center")해당 시각화 테이블 형성 코드
위의 테이블에서 확인할 수 있듯, train_clinical_data에서 1개의 null value를 가지는 항목은 updrs_3, updrs_4, upd23b_clinical_state_on_medication으로 특히 upd23b_clinical_state_on_medication 부분에서 null value가 높은 빈도로 존재함을 알 수 있다. 해당 항목은 환자의 약물 상태에 대한 기록으로 어떻게 처리할지가 중요하다. updrs_3은 운동성 검사에 대한 점수이며, updrs_4는 운동성 합병증에 대한 검사이므로 이는 파킨슨병의 주요 증상을 나타내는 지표이기 때문에 여전히 중요한 항목이다. 이 점수는 0에 가까울수록 정상으로 판단하므로 null values를 0으로 보고 처리할 수 없다는 점이 중요하다.
2개의 null values를 가지는 항목은 updrs_4, upd23b_clinical_state_on_medication라고 볼 수 있다.
3개의 null values를 가지는 항목은 updrs_3, updrs_4, upd23b_clinical_state_on_medication으로 3가지 모두 높은 null values 빈도를 가짐을 확인할 수 있다.
4개의 null values를 가지는 항목은 updrs_1, updrs_2, updrs_4, upd23b_clinical_state_on_medication이다.
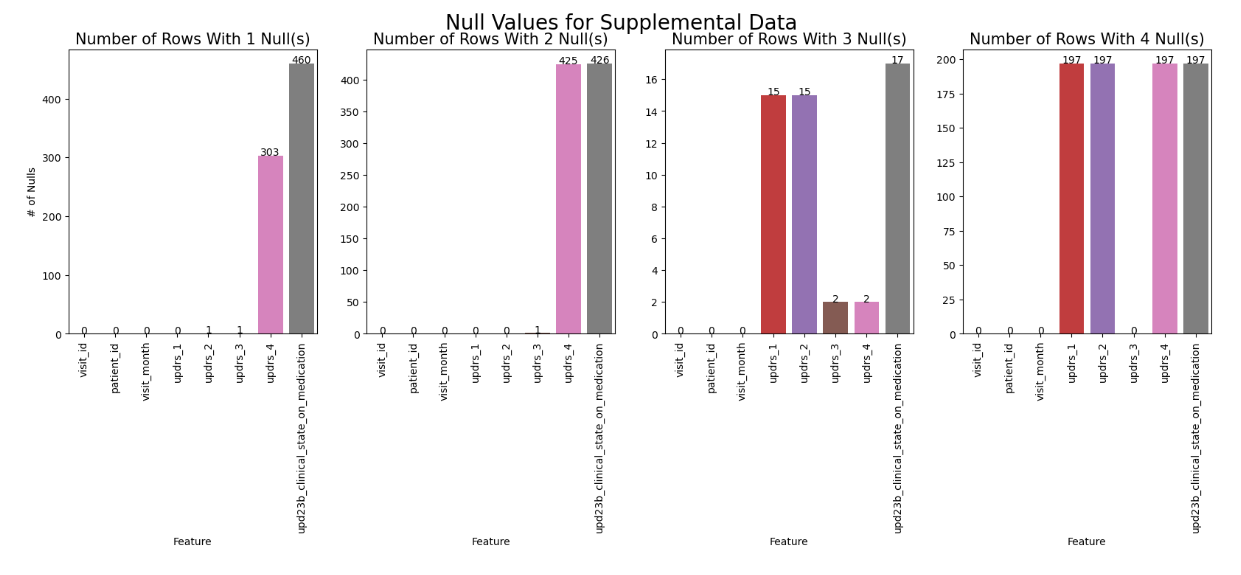
null_count_labels = [supplemental_clinical_data[(supplemental_clinical_data["null_count"] == x)].isnull().sum().index[:-1] for x in range(1, 6)]
null_count_values = [supplemental_clinical_data[(supplemental_clinical_data["null_count"] == x)].isnull().sum().values[:-1] for x in range(1, 6)]
fig, axs = plt.subplots(nrows=1, ncols=4, figsize=(20, 5))
fig.suptitle("Null Values for Supplemental Data", fontsize=20)
axs = axs.flatten()
for x in range(0, 4):
ax = axs[x]
labels = null_count_labels[x]
_ = sns.barplot(x=labels, y=null_count_values[x], ax=ax)
_ = ax.set_title("Number of Rows With {} Null(s)".format(x + 1), fontsize=15)
_ = ax.set_ylabel("# of Nulls" if x == 0 else "")
_ = ax.set_xlabel("Feature")
_ = ax.set_xticks([z for z in range(len(labels))], labels, rotation=90)
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:d}".format(int(height)), ha="center")해당 시각화 테이블 형성 코드
앞선 train_clinical_data와 전반적으로 비슷한 양상을 띄며 3개의 null values가 있는 경우는 약간 차이가 있는 모습을 확인할 수 있다.
다음과 같은 상황에서 앞으로 null values를 어떻게 처리할지가 중요한 과제가 될 것임을 알 수 있다.
1.3 Duplicated Rows

titles = ["Peptide Data", "Protein Data", "Clinical Data", "Supplemental Data"]
value_counts = []
duplicates = train_peptides.pivot_table(index=[
'UniProt', 'Peptide', 'PeptideAbundance',
], aggfunc="size")
unique, counts = np.unique(duplicates, return_counts=True)
value_counts.append(dict(zip(unique, counts)))
duplicates = train_protiens.pivot_table(index=[
'UniProt', 'NPX',
], aggfunc="size")
unique, counts = np.unique(duplicates, return_counts=True)
value_counts.append(dict(zip(unique, counts)))
duplicates = train_clinical_data.pivot_table(index=[
'visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4', 'upd23b_clinical_state_on_medication'
], aggfunc="size")
unique, counts = np.unique(duplicates, return_counts=True)
value_counts.append(dict(zip(unique, counts)))
duplicates = supplemental_clinical_data.pivot_table(index=[
'visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4', 'upd23b_clinical_state_on_medication'
], aggfunc="size")
unique, counts = np.unique(duplicates, return_counts=True)
value_counts.append(dict(zip(unique, counts)))
fig, axs = plt.subplots(nrows=1, ncols=4, figsize=(20, 4))
axs = axs.flatten()
for x in range(4):
ax = axs[x]
_ = sns.barplot(x=list(value_counts[x].keys())[1:], y=list(value_counts[x].values())[1:], ax=ax)
_ = ax.set_title("Duplicate Counts in {}".format(titles[x], fontsize=15))
_ = ax.set_ylabel("Count")
_ = ax.set_xlabel("Number of Times Row is Duplicated")
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:d}".format(int(height)), ha="center")해당 시각화 테이블 형성 코드
Peptide data 에서 1,765개 행이 두 번 중복되어 나타났고, 2개 행이 세 번 중복되어 나타났다. 중복도는 전체의 0.36%이다.
Protein data 에서 400개 행이 두 번 중복되어 나타났다. 중복도는 전체의 0.35%이다.
Clinical data 에서 2개 행이 두 번 중복되어 나타났다. 중복도는 전체의 0.15%이다.
Supplemental data 에서 7개 행이 두 번 중복되어 나타났다. 중복도는 전체의 0.63%이다.
이를 통해 2종의 임상 dataset의 중복값은 크게 문제되지 않을 것으로 보이며, 펩타이드와 단백질 dataset의 경우 중복값이 어느 정도 영향을 끼칠 수 있을 것으로 확인된다.
1.4 - Statistical Breakdown
- Clinical vs Supplemental Data
features = [
'visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4',
]
train_clinical_data[features].describe().T.style.bar(subset=['mean'], color='#7BCC70')\
.background_gradient(subset=['std'], cmap='Reds')\
.background_gradient(subset=['50%'], cmap='coolwarm')clinical data 정보 확인
features = [
'visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4',
]
supplemental_clinical_data[features].describe().T.style.bar(subset=['mean'], color='#7BCC70')\
.background_gradient(subset=['std'], cmap='Reds')\
.background_gradient(subset=['50%'], cmap='coolwarm')supplemental data 정보 확인

train_clinical_data["origin"] = "Clincial Data"
supplemental_clinical_data["origin"] = "Supplemental Data"
combined = pd.concat([train_clinical_data, supplemental_clinical_data]).reset_index(drop=True)
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(15, 5))
sns.set_style('darkgrid')
_ = sns.histplot(data=combined, x="visit_month", hue="origin", kde=True, ax=ax, element="step")
_ = ax.set_title("Visit Month by Data Source", fontsize=15)
_ = ax.set_ylabel("Count")
_ = ax.set_xlabel("Visit Month")해당 시각화 테이블 형성 코드
방문 기간에 따른 KDE 그래프로 clinical data는 주로 0~36정도에서 나타나는 반면, supplemental data는 0~108정도까지 나타남을 알 수 있다. 이때, KDE란, kernel density estimation로 통계학에서 확률 밀도 추정을 위한 kernel smoothing의 적용, 즉 가중치를 통해 kernel을 기반으로 random variable의 확률 밀도 함수를 추정하는 비모수 방법이다. (참고 : https://en.wikipedia.org/wiki/Kernel_density_estimation)
또한, updrs_1, updrs_2, updrs_3, updrs_4를 비교해 보았을 때(해당 그래프는 본 페이지 참고), updrs_1, updrs_4는 비슷한 양상을 띠었지만 updrs_2, updrs_3의 경우 supplemental data가 0에 근접하는 점수의 빈도가 더 높음을 확인할 수 있었다.
다음으로 adversarial validation을 수행하여 clinical data와 supplemental data 사이의 차이를 대략적으로 측정할 수 있다. ROC AUC score를 통해 두 data의 차이를 확인할 수 있으며 0.5에 가까울수록 두 data가 유사하고 1에 가까울수록 차이가 확실하다.

from sklearn.model_selection import KFold
from sklearn.metrics import roc_auc_score
from lightgbm import LGBMClassifier
from lightgbm import early_stopping, log_evaluation
from sklearn.metrics import confusion_matrix
from sklearn.metrics import roc_curve
from sklearn.preprocessing import LabelEncoder
train_clinical_data["origin"] = 0
supplemental_clinical_data["origin"] = 1
combined = pd.concat([train_clinical_data, supplemental_clinical_data]).reset_index(drop=True)
features = [
'visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4', 'upd23b_clinical_state_on_medication',
]
le = LabelEncoder()
combined['upd23b_clinical_state_on_medication'] = le.fit_transform(combined['upd23b_clinical_state_on_medication'])
n_folds = 5
skf = KFold(n_splits=n_folds, random_state=2023, shuffle=True)
train_oof_preds = np.zeros((combined.shape[0],))
train_oof_probas = np.zeros((combined.shape[0],))
for fold, (train_index, test_index) in enumerate(skf.split(combined, combined["origin"])):
print("-------> Fold {} <--------".format(fold + 1))
x_train, x_valid = pd.DataFrame(combined.iloc[train_index]), pd.DataFrame(combined.iloc[test_index])
y_train, y_valid = combined["origin"].iloc[train_index], combined["origin"].iloc[test_index]
x_train_features = pd.DataFrame(x_train[features])
x_valid_features = pd.DataFrame(x_valid[features])
model = LGBMClassifier(
random_state=2023,
objective="binary",
metric="auc",
n_jobs=-1,
n_estimators=2000,
verbose=-1,
max_depth=3,
)
model.fit(
x_train_features[features],
y_train,
eval_set=[(x_valid_features[features], y_valid)],
callbacks=[
early_stopping(50, verbose=False),
log_evaluation(2000),
]
)
oof_preds = model.predict(x_valid_features[features])
oof_probas = model.predict_proba(x_valid_features[features])[:,1]
train_oof_preds[test_index] = oof_preds
train_oof_probas[test_index] = oof_probas
print(": AUC ROC = {}".format(roc_auc_score(y_valid, oof_probas)))
auc_vanilla = roc_auc_score(combined["origin"], train_oof_probas)
fpr, tpr, _ = roc_curve(combined["origin"], train_oof_probas)
print("--> Overall results for out of fold predictions")
print(": AUC ROC = {}".format(auc_vanilla))
confusion = confusion_matrix(combined["origin"], train_oof_preds)
fig, axs = plt.subplots(nrows=1, ncols=2, figsize=(15, 8))
labels = ["Clincial Data", "Supplemental Data"]
_ = sns.heatmap(confusion, annot=True, fmt=",d", ax=axs[0], xticklabels=labels, yticklabels=labels)
_ = axs[0].set_title("Confusion Matrix (@ 0.5 Probability)", fontsize=15)
_ = axs[0].set_ylabel("Actual Class")
_ = axs[0].set_xlabel("Predicted Class")
_ = sns.lineplot(x=[0, 1], y=[0, 1], linestyle="--", label="Indistinguishable Datasets", ax=axs[1])
_ = sns.lineplot(x=fpr, y=tpr, ax=axs[1], label="Adversarial Validation Classifier")
_ = axs[1].set_title("ROC Curve", fontsize=15)
_ = axs[1].set_xlabel("False Positive Rate")
_ = axs[1].set_ylabel("True Positive Rate")해당 시각화 테이블 형성 코드
위의 곡선에서 확인하였듯, clinical data와 supplemental data는 구조가 다르므로 두 dataset을 결합할 때 주의를 요함을 알 수 있다.
추가적으로 해당 페이지에서는 peptide data와 protein data에 대한 KDE 그래프를 시각화하였으며 이를 통해 이 data들은 넓은 범위에 분포하여 존재하기 때문에 세부적으로 분류하여 분석해야함을 알 수 있었다.
2. Feature Exploration
방문 기간은 다양한 dataset에 영향을 미친다.
- Visit Month vs UPDRS
UPDRS 점수 관찰을 약물을 복용하는 그룹과 복용하지 않는 그룹으로 세분화해야 한다. 이 탐색의 목적상 약물 상태와 관련된 임상 데이터에서 발견된 null 값은 Off로 간주하였다(앞으로 이 부분에 대해 논의해봐야 할 것이다).
약물을 사용한(On values) 경우, 각 UPDRS 점수와 각 방문 기간에 걸쳐 많은 양의 분산과 특이치가 있으며 일반적으로 UPDRS part 1, 2, 4에서 점수의 추세선은 비교적 평평하게 유지된다. UPDRS part 3에서는 점수가 점진적으로 증가한다.
Holden et al(2018)에서 UPDRS의 최대 점수는 272점이라고 언급한다. 이전 버전의 UPDRS에서는 시간이 지남에 따라 UPDRS 점수가 선형적으로 진행되었다. 이 데이터 내에서 시간이 지남에 따라 점수가 증가하는지 확인하기 위해 UPDRS 점수의 합계를 살펴보았다.
약을 끊는 동안 모든 UPDRS 점수의 합계로 방문 기간이 증가함에 따라 상승 추세를 확인할 수 있으며, 이는 전체적으로 질병 진행이 발생하고 있음을 나타낸다. 의약품을 사용하는 동안 추세선은 96개월 이상까지 비교적 평탄한 상태를 유지하며 전체 점수가 증가하고 이것은 질병의 진행이 일어나고 있음을 나타낸다.

train_clinical_data["updrs_sum"] = train_clinical_data["updrs_1"] + train_clinical_data["updrs_2"] + train_clinical_data["updrs_3"] + train_clinical_data["updrs_4"]
train_clincial_data_copy = train_clinical_data.copy()
train_clincial_data_copy["upd23b_clinical_state_on_medication"] = train_clincial_data_copy["upd23b_clinical_state_on_medication"].fillna("Off")
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(15, 5))
sns.set_style('darkgrid')
_ = sns.boxplot(data=train_clincial_data_copy, x="visit_month", y="updrs_sum", ax=ax)
_ = sns.pointplot(data=train_clincial_data_copy, x="visit_month", y="updrs_sum", color="r", ci=None, linestyles=[":"], ax=ax)
_ = ax.set_title("UPDRS Sum of Scores by Month", fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("Score")해당 시각화 테이블 형성 코드
전반적으로, UPDRS 점수가 증가하는 추세를 볼 수 있다. 이 관찰은 점수가 감소하기보다는 시간이 지남에 따라 증가한다는 것을 시사하기 때문에 중요하다. 이것은 기계 학습 알고리즘의 예측이 타당한지 확인하기 위한 사후 처리 검사로 사용될 수 있다고 한다.
Visit Month vs Protein Data
매달 범주에 걸쳐 안정적인 양의 단백질 발현을 볼 수 있다. 단백질 발현 빈도와 관련하여 큰 차이가 있지만, 전체적으로 동일한 평균 총 NPX 값이 몇 달 동안 반복되는 것을 볼 수 있다. 이것은 절대적인 숫자보다는 실제 발현되는 단백질에 차이가 있을 것이라는 것을 의미한다.
227개의 모든 단백질을 보는 것은 어려우므로 몇 달 동안 현저하게 증가하거나 감소하는 단백질을 살펴볼 것이다. 227개의 모든 단백질에 대한 단백질 수를 조사한 다음 평균에 비해 표준 편차가 매우 큰 것으로 보이는 것을 선택할 것이다. 이 페이지의 EDA에서는 표준 편차가 평균 값의 25% 이상인 단백질 발현 데이터를 확인할 수 있었다.

unique_proteins = train_protiens["UniProt"].unique()
unique_months = train_protiens["visit_month"].unique()
protein_dict = dict()
for protein in unique_proteins:
if protein not in protein_dict:
protein_dict[protein] = {
"months": unique_months,
"count_NPX": [train_protiens[(train_protiens["UniProt"] == protein) & (train_protiens["visit_month"] == month)]["NPX"].count() for month in unique_months],
"total_NPX": [train_protiens[(train_protiens["UniProt"] == protein) & (train_protiens["visit_month"] == month)]["NPX"].sum() for month in unique_months],
"avg_NPX": [0 * len(unique_months)],
}
for protein in unique_proteins:
protein_dict[protein]["avg_NPX"] = [float(total) / count for total, count in zip(protein_dict[protein]["total_NPX"], protein_dict[protein]["count_NPX"])]
for protein in unique_proteins:
protein_dict[protein]["min_NPX"] = min(protein_dict[protein]["avg_NPX"])
protein_dict[protein]["max_NPX"] = max(protein_dict[protein]["avg_NPX"])
for protein in unique_proteins:
protein_dict[protein]["mean"] = sum(protein_dict[protein]["avg_NPX"]) / len(protein_dict[protein]["months"])
protein_dict[protein]["std"] = sum([(total_NPX - protein_dict[protein]["mean"]) ** 2 for total_NPX in protein_dict[protein]["avg_NPX"]]) / (len(unique_months) - 1)
protein_dict[protein]["std"] = protein_dict[protein]["std"] ** 0.5
proteins_with_large_std = [protein for protein in unique_proteins if protein_dict[protein]["std"] > (protein_dict[protein]["mean"] * .25)]import math
proteins_of_interest_by_month = {
"UniProt": [],
"Visit Month": [],
"Average NPX": [],
}
for protein in proteins_with_large_std:
for month_index, month in enumerate(unique_months):
proteins_of_interest_by_month["UniProt"].append(protein)
proteins_of_interest_by_month["Visit Month"].append(month)
value = protein_dict[protein]["avg_NPX"][month_index]
value /= protein_dict[protein]["max_NPX"]
proteins_of_interest_by_month["Average NPX"].append(value)
df = pd.DataFrame(proteins_of_interest_by_month)
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(20, 8))
sns.set_style('darkgrid')
_ = sns.lineplot(data=df, x="Visit Month", y="Average NPX", hue="UniProt", style="UniProt", ax=ax)
_ = ax.set_title("Average NPX per Protein by Month", fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("Average NPX")
_ = plt.legend(ncol=5)해당 시각화 테이블 형성 코드
위의 그래프를 통해 알 수 있는 사실은 우선, 30개월, 54개월, 96개월에 관심 있는 모든 단백질의 평균 수준이 상향 및 하향으로 급상승하는 것을 볼 수 있다. 이것이 약물 상태나 UPDRS 점수와 같은 임상 데이터와 상관관계가 있는지 여부는 단백질 특징을 직접 살펴볼 때 더 자세히 살펴볼 것이다. 단백질과 방문 기간 사이에 상관관계는 있어보인다.
다음으로 여러 단백질이 위쪽과 아래쪽으로 모두 이동한다. 이것은 단백질이 UPDRS 점수와 양의 또는 음의 상관관계를 가질 수 있음을 의미한다.
마지막으로 월별 차이가 매우 큰 단백질 발현 빈도(표준 편차 > 평균의 25%)만 보고 있다는 것이다. 기계 학습 알고리즘이 학습할 수 있는 표현 빈도의 더 미묘한 변화가 있을 수 있다.
2. 추가 아이디어
해당 페이지 이외에도 공유되어있는 여러 코드들과 EDA들을 보면서 F1 score 와 AUC ROC score의 개념을 적용해보는 아이디어를 떠올릴 수 있었다.
F1 score란 재현율과 정밀도의 개념과 밀접한 관련이 있는 점수로,
정밀도는 확실한 기준이 되는 경우만 positive로 설정하고 나머지는 negative로 설정하려할수록 100% 가까운 값을 가지고,
재현율을 모든 경우를 positive로 설정하려할 경우 100%의 값을 가진다.
F1 score는 이러한 정밀도와 재현율을 결합한, 즉 정밀도와 재현율의 조화평균 값이다.
우리는 null values를 처리하는 과정에서 전부 0으로 처리하거나 제외할 수 없는 상황이므로 최대한 관계성을 찾아 값을 지정하여 처리해야 하는데 이때 F1 score를 통해 그 정도를 조절하며 모델의 정확도를 높일 수 있을 것으로 예상된다.


구체적인 공식과 F score에 대한 정보는 https://en.wikipedia.org/wiki/F-score를 참고할 예정이다.
또한 ROC AUC curve는 의학 분야와 머신러닝 이진 분류 모델의 예측 성능 판단에 많이 사용되는 평가 지표이다. 해당 곡선의 개념과 공식은 https://en.wikipedia.org/wiki/Receiver_operating_characteristic를 참고할 예정이다.
이로써 전반적인 EDA 결과와 아이디어를 얻을 수 있었다. 다음에는 직접 EDA 코드를 작성해볼 것이다.
'공모전' 카테고리의 다른 글
| AMP®-Parkinson's Disease Progression Prediction 공모전 (3) (0) | 2023.05.20 |
|---|---|
| AMP®-Parkinson's Disease Progression Prediction 공모전 (1) (0) | 2023.03.12 |

